Delivering Less Expensive and Higher Quality sequencing with Reduced Turn-Around-Times
Based on your needs, experts determine the depth and coverage required
Experts can review your project design and sample selection criteria.
We offer Next Generation Sequence services on
different NGS platforms depending upon the type of sample and required sequencing depths.
DNA
Whole genome sequencing
Whole-genome sequencing is the most comprehensive method for analyzing the genome. It provides a high-resolution, base-by-base view of the whole genome. It can capture both common and rare variants that might otherwise be missed. It is ideal for discovery applications, such as identifying causative variants and novel genome assembly. Whole-genome sequencing can detect single nucleotide variants, insertions/deletions, copy number changes, and large structural variants.
Whole exome sequencing
Exome sequencing is one of the most widely used comprehensive targeted sequencing methods. The exome (protein-coding) represents less than 2% of the genome, but it is estimated to contain ~85% of the known disease-related variants. This makes whole-exome sequencing a cost-effective alternative to whole-genome sequencing.
Custom Targeted sequencing
Targeted sequencing, allows isolating and sequencing a subset of genes or regions of the genome. This helps us to focuses on regions of interest, generating smaller and more manageable data sets. Also reduces the sequencing costs and data analysis burdens. Reduces turnaround time and enables deep sequencing at high coverage levels for rare variant identification.
Microbe and Microbiome sequencing
NGS enables fast, accurate characterization of any species. Experimental strains, genomes with high plasticity which involves bacterium, virus, or other microbe. Sequencing small microbial genomes can be useful for contaminated foods, infectious disease and epidemiology studies etc.
Human microbiome means the microbes that are found in and on the human body. The goal of human microbiome sequencing studies is to understand the role of microbes in health and disease.
De novo sequencing
We also sequence unknown genomes and genomes with no reference sequence. De novo sequencing refers to sequencing a novel genome where there is no reference sequence available.
RNA
Reference RNA sequencing
RNA-Seq is highly sensitive and accurate tool for measuring expression across the transcriptome. It allows researchers to detect both known and novel transcript isoforms, gene fusions, single nucleotide variants, allele-specific gene expression and other features without the limitation of prior knowledge. It enables more sensitive and accurate measurement of gene expression compare to traditional methods and can be applied to any species. The price per sample turns out to be lower than many arrays.
Small RNA sequencing
Small RNA sequencing (RNA-Seq) is a technique to isolate and sequence small RNA species, such as microRNAs (miRNAs). With small RNA-Seq you can discover novel miRNAs and other small noncoding RNAs, and examine the differential expression of all small RNAs in any sample. We can understand how post-transcriptional regulation contributes to phenotype. It is also possible to identify novel biomarkers.
Epigenomes sequencing
Bisulfite- sequencing
With whole genome bisulfite sequencing, it is possible to view methylation at practically every cytosine in the genome across most species with small amounts of DNA. With targeted methylation sequencing, we can focus on regions of interest and reduce the sequencing costs and turnaround time.
Chip- sequencing
ChIP-sequencing, also known as ChIP-seq, is a method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins.
NGS Data Quality
Before the data is analyzed, it is crucial to check the quality of the data. Our Bioinformaticians take the lead role in NGS data quality control. Standard tool are used for checking the quality of data generated on Illumina and other NGS platforms.
Basic statistics
» Per base sequence quality
» Per sequence quality scores
» Per base sequence content
» Per sequence GC content
» Per base N content
» Sequence Length Distribution
» Sequence duplication levels
» Over-represented sequences
» Adapter/Kmer content
The NGS platforms generate millions of reads per sample. How to efficiently and correctly map these millions of short reads to a reference genome is one of the major challenges in NGS data analysis.
To align your Next-Generation sequencing data to the reference, your reads should be in FastA or FastQ format. Most sequencers have their own native formats, unless the data is converted into FastA or FastQ format, most of the available aligners will not be able to align the reads.
Based on the project (DNA, RNA or bisulfite Seq) a different aligner has to be employed. Our Bioinformaticians convert the data to the required format, and pick the right aligner for you.
DNA
» Whole genome assembly
» Whole exome assembly
» De-novo genome assembly
» Targeted sequence assembly
RNA
» Reference RNA-Seq assembly
» miRNA-Seq assembly
» De-novo RNA-Seq assembly
Epigenomes assembly
» Bisulfite-seq assembly
» Chip-seq assembly
NGS Data Analysis
Our Bioinformaticians use novel computational methods to discover, catalogue, and characterize genomic variation form DNA, RNA and Methyl Seq data sets.
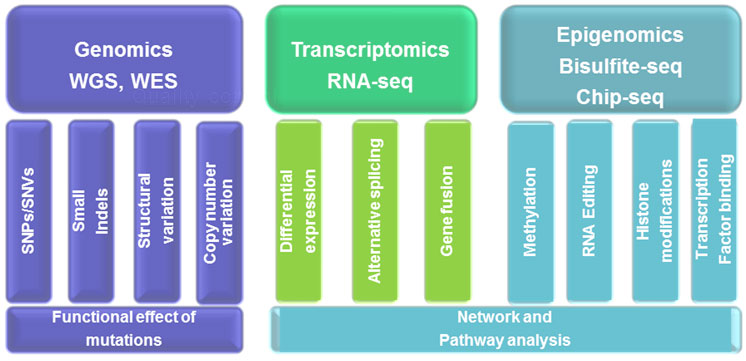
Genomics
Whole Genome, Exome and Targeted Sequence Data Analysis
Advanced analysis and visual analytics for DNA Sequencing.
Rapid analysis of raw DNA-seq reads to identify single nucleotide polymorphisms and structural rearrangements such as insertions, deletions, duplications, copy number variation, inversions and translocations.
Analysis for single genomes, trios, cohorts, or tumor-normal pairs.
Transcriptomics
RNA and miRNA Seq Data Analysis
Comprehensive gene expression profiling and analysis.
Rapid analysis of raw RNA-seq or miRNA seq reads to gain insights into target genes, and overall expression levels.
Comprehensive analysis of upregulated and down regulated genes
Differentially expressed genes across samples, time series, treatments or any condition or criteria
Epigenomics
Methyl and Chip-Seq Data Analysis
Genomic data is never interpreted in isolation. How are you integrating patient records or clinical research with�clinical diagnostics development? Hospital, university, drug development � all can benefit from different pieces of the same knowledge base. omnomicsNGS has been designed to be easily integrated into large IT infrastructures.
Request more information, a webinar, or a visit? Get in Touch